
Help on
Search Guidelines
- name: provide a descriptive name for your search. This name will be used for the output files to download so that you can keep track of different searches.
- Select either single query or batch search: in the first case, the sequence can be up to 500 nucleotides long. Paste a plain sequence, not fasta format. Only characters representing valid nucleotides (A,a,C,c,G,g,T,t,N,n) will be considered and any other character will be discarded. In the second case you can provide a (multi-)fasta file with any number of sequences and a total size of up to 500KB.
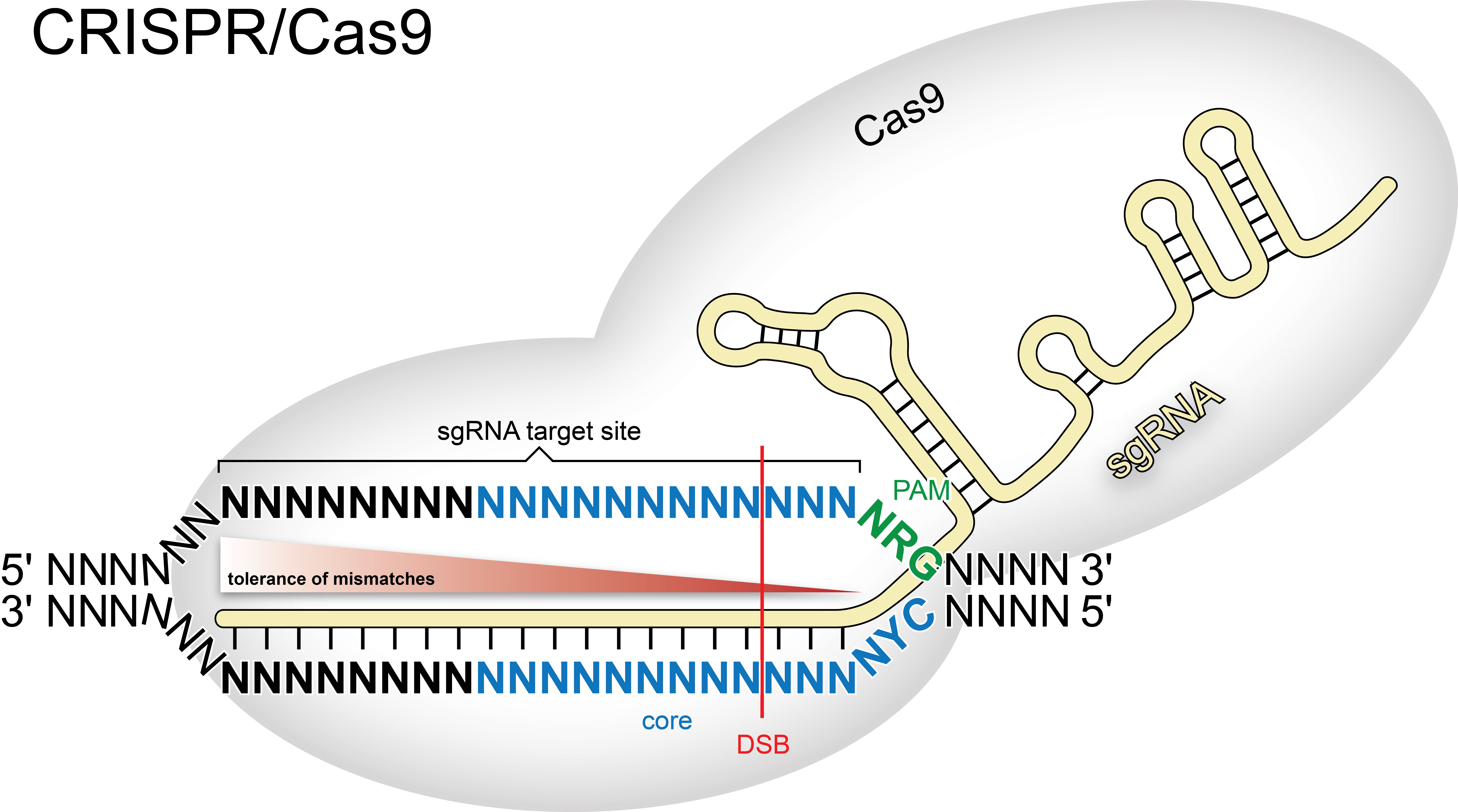
- PAM type: apart from the protospacer adjacent motif (PAM) recognized by the Cas9 protein of Streptococcus pyogenes (SP), other motifs have been identified for different bacterial species. In addition, it has been shown that the Cas9 endonuclease in SP can also cleave sgRNA target sites followed by ‘NAG’, however with efficiency reduced to ∼20% [Hsu et al.], rendering a PAM motif ‘NRG’. Selecting a PAM motif other than the one recognized by the Cas9 of SP will disable the use of the core parameters for off-target site search (see below). We also include the PAM of the Cpf1 endonuclease [Zetsche et al.], from Acidaminococcus or Lachnospiraceae, that recognizes a 'TTTN' motif.
Target selection
- target site length: the length of the sgRNA target site, excluding the PAM sequence, can be from 15 to 23 bases.
- target site 5’ limitation: typical in vitro transcription promoters, like the T7 or the U6, require one or two leading ‘G’ respectively. However, we and others [Hwang et al., Ansai & Kinoshita] found that adding one or two Gs to the desired target sequence can also give valid sgRNAs that induce double strand breaks (DSBs). This release of restriction identifies many more target sites in the query sequence (hence ‘NN’ is the default value).
- target site 3’ limitation: for C. elegans it has been shown that ‘GG’ at the 3’ site enhances the introduction of a DSB [Farboud & Meyer]. However, as with the previous field, you can specify any valid sequence of two bases to restrict the list of possible CRISPR/Cas9 target sites.
- in vitro transcription: Select your in vitro transcription method. We will provide the forward and reverse oligos to clone each candidate sgRNA into the appropriate vector. If you select "Custom" you can specify your overhangs, which will be appended to the 5' of the repective primers.
Off-target prediction
- max. total mismatches: can be set to 0-5. Note: more than four mismatches in total prevent DSB induction [Hsu et al., Cho et al.].
- max. core length: Mismatches at a distance to the PAM, will still allow the introduction of a DSB, while mismatches close to the PAM will abolish the introduction of a DSB [Hsu et al., Cong et al., Stemberg et al.]. The core is a simplified parameter to account for these findings and is defined as the nucleotides adjacent to the PAM (12 by default, can be set to 2-20). The checkbox in front allows to enable/disable the core parameters. The prediction of off-target sites taking into account the core is only available when using the PAM motif from S. pyogenes. In the other cases there is not yet experimental evidence suggesting that the core can be benefitial.
- max. core
mismatches: can be set to 0-2. Note: in the core, more than 2
mismatches abolishes DSB introduction at the potential off-target
position [Hsu et al., Cong et al.]
- species: define in which genomic context off-targets should be predicted.
Output
Single query
During search, a page will auto refresh indicating how many candidate sgRNAs were identified. After the search process, you will automatically be forwarded to the results page. In the following example a genomic sequence from human TP53 gene was used.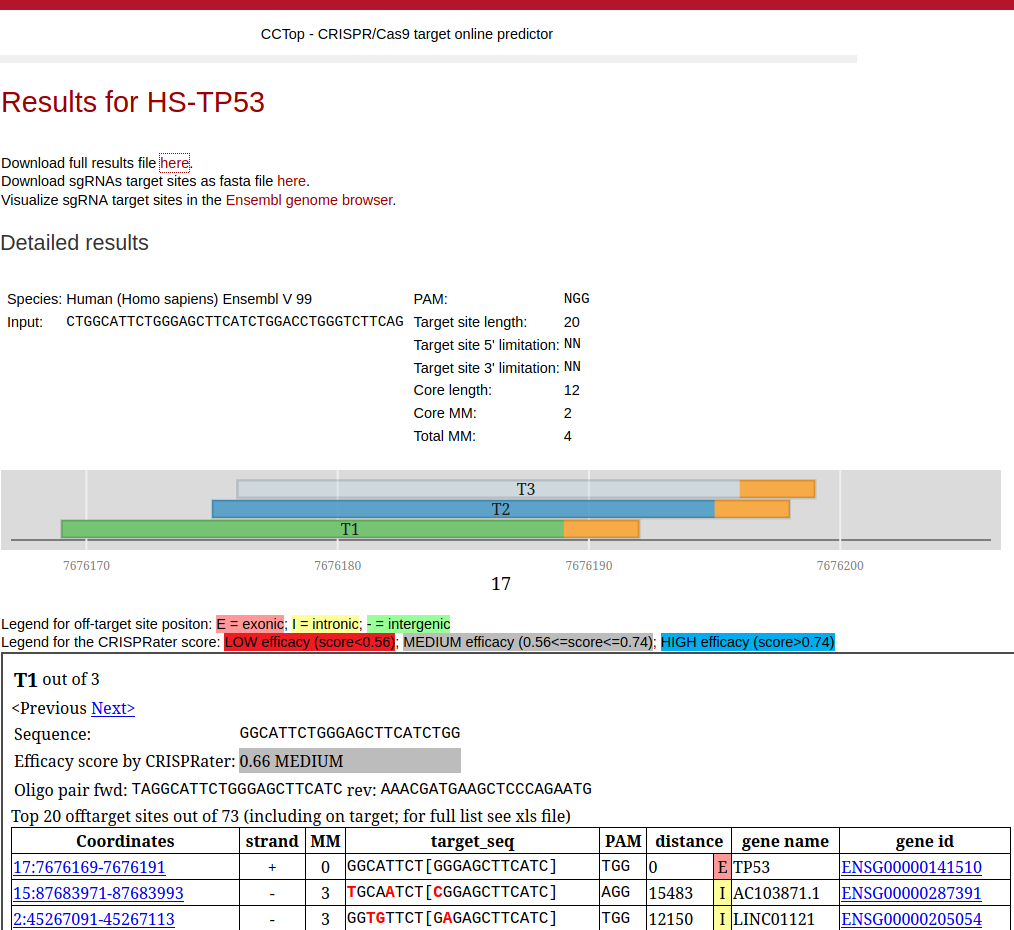
- to download the full results file (tab separated) like shown in the table at the bottom.
- to download a fasta file containing all identified sgRNA target sites
- to visualize the locus of the query sequence in the Ensembl browser with the identified sgRNA target sites, whenever the query sequence belogs to the selected as targeted genome and this specues was added to CCTop from Ensembl. A similar link will be available for the UCSC browser
A graphical representation of the query sequence with the identified sgRNA target sites (colored by score, see below) as well as a full list of all candidates is given ranked by taking into account the number of total off-target sites, the distribution of mismatches and the proximity to exons. It is possible to click on any of the displayed target sites to focus the list below on the output corresponding to this site. For each sgRNA target site it is shown the predicted efficacy score by CRISPRater [Labuhn et al.], with the color code shown above, together with cloning oligonucleotides (5’-3’ orientation) depending om the in vitro transcription method selected. For the T7 promoter, if the candidate sgRNA sequence does not start with two Gs, the sequence is extended or the initial bases are changed to obtain the required two Gs at the 5’ end. The extended or substituted bases are given in small case for recognition. For the U6 promoter the same procedure is taken only that in this case only one G at the 5’ end is required. In other case, the given overhang sequence is appended to the identified sgRNA target sequence.
Detailed information is provided for each potential off-target site (only at most 20 are shown, for the full list refer to the .xls file):
- Coordinates: with link to the corresponding browser, if applicable
- strand: orientation of the (off-) target site
- MM: number of mismatches
- target_seq: off-target sequence with highlighted mismatches in red, core in square brackets
- PAM: endogenous PAM of the (off-) target site
- distance: distance to the closest exon (0 if target site and exon coordinates overlap; NA for target sites farther than 100kb to the next exon). Further information on the location of the off-target site is provided by a colour code: green = intergenic; yellow = intronic; red = exonic.
- gene name: the corresponding gene name
- gene id: the corresponding gene id (with link to Ensembl, if applicable) and identifier.
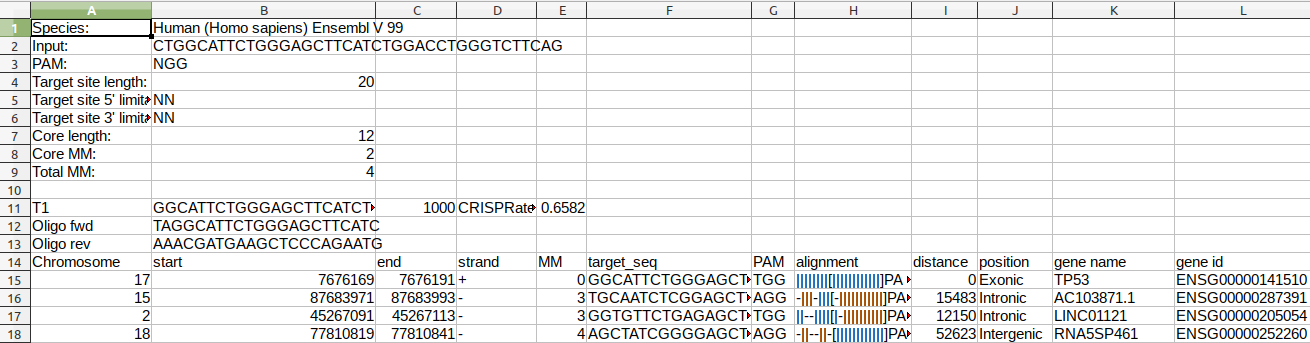
Each candidate (target) is labelled with the prefix "T" and a correlative number from 1 to the number of candidates. Candidates are scored from 1000 - suggested best choice to 0 - worst choice. This score takes into account the number of off-targets in the genome, their quality, i.e. number of mismatches and position with respect to the PAM, and the distance to gene exons. The off-target sites for each target site are internally ranked by decreasing likelihood of potential Cas9 activity. If the query sequence is derived from the same genome against which the off-target sites were predicted, the first hit of each target is the candidate target itself, displaying its properties.
Batch query
The result page for a batch search is slightly different. During the search process the page indicates which sequence is being analysed and the refresh time is longer. For this kind of tasks it is advisable to provide a valid email address so that when the search is finished this will be notified with a message to that address.Once the search process is finished successfully the result page will offer a link to download the full set of results in an archive with zip format. Also a link to the specific result page, as described for a single query, will be given for each one of the sequences contained in the input fasta file.
The content of the zip archive consists of the bed, fasta and xls file described above and a html file to visualize locally the results in a web browser. Note that for batch searches only a maximum of 50 off-target sites will be considered, if you need the exahustive list of off-target sites you can later run a searach in single sequence mode with the target site of interest.
References
- Ansai, S. & Kinoshita, M. Targeted mutagenesis using CRISPR/Cas system in medaka. Biol. Open 3, 362–71 (2014).
- Cho, S. W. et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24, 132–41 (2014).
- Cong, L. et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science (2013). doi:10.1126/science.1231143
- Farboud B. & Meyer B. J. Dramatic Enhancement of Genome Editing by CRISPR/Cas9 Through Improved Guide RNA Design. Genetics (2015).
- Hsu, P. D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–32 (2013).
- Hwang, W. Y. et al. Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PLoS One 8, e68708 (2013).
- Labuhn, M. et al. Refined sgRNA efficacy prediction improves large- and small-scale CRISPR–Cas9 applications. Nucleic Acids Research. 46(3) 1375-1385 (2017).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C. & Doudna, J. a. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–7 (2014).
- Zetsche, B. et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell (2015)
Access to the results
The results produced by CCTop will be available online for a period of time depending on the type of search after the seach job has finished, either successfully or not:
- Single search: 48 hours
- Batch search: 7 days
Citing this tool
If you use this tool for you scientific work, please cite it as:
Stemmer,
M., Thumberger, T., del Sol Keyer, M., Wittbrodt, J. and Mateo,
J.L. CCTop: an intuitive, flexible and reliable CRISPR/Cas9
target prediction tool. PLOS ONE (2015). doi:
10.1371/journal.pone.0124633
If you use the CRISPRater score to select your target sites please cite as well this work:
Labuhn,
M., Adams, F. F., Ng, M., Knoess, S., Schambach, A., Charpentier, E. M., … Heckl, D.
Refined sgRNA efficacy prediction improves large- and small-scale CRISPR–Cas9 applications.
Nucleic Acids Research (2017). doi: 10.1093/nar/gkx1268
Recommendation:
Until now there have been many different computational tools to predict sgRNA efficacy. We offer one of those that is CRISPRater. However, these predictions are yer far from being perfect and there are many factors that can influence the final efficacy of a CRISPR experiment. Therefore, we recommend check carefully the results of the experiments in a independent manner.Feedback:
If you encounter any problem using this tool or you have any suggestion, please email us:ed.grebledieh-inu.soc@rpsirc.